FDA’s Division of Nonprescription Drug Products
By Mark Land, AAHP president
August 1, 2017
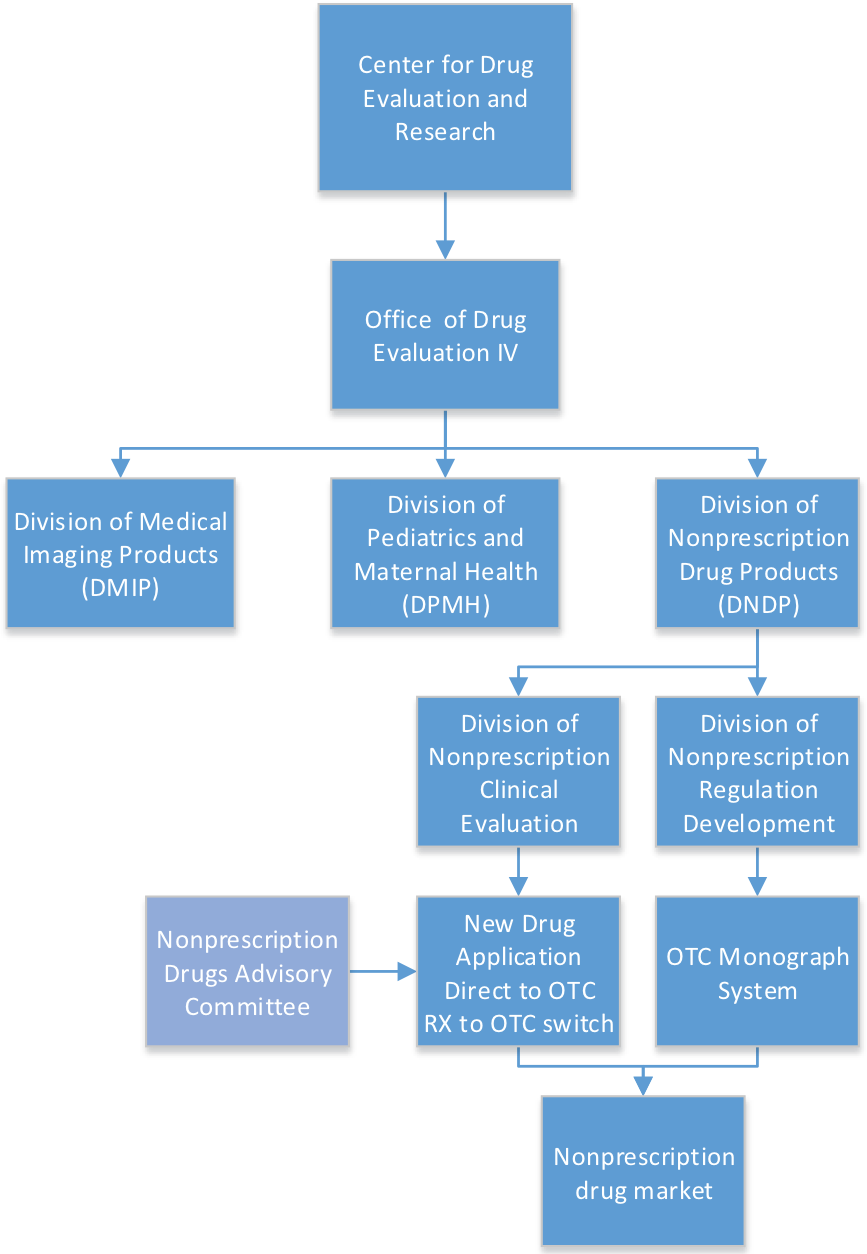 The development and regulation of over-the-counter (OTC) drug products is the responsibility of the Division of Nonprescription Drug Products (DNDP) within the Center for Drug Evaluation and Research’s (CDER) Office of Drug Evaluation IV. A sponsor seeking to market its product OTC, either as a new drug application (NDA) or as a switch from a prescription product, applies to the DNDP, which is also responsible for the development of the OTC drug monographs.
The development and regulation of over-the-counter (OTC) drug products is the responsibility of the Division of Nonprescription Drug Products (DNDP) within the Center for Drug Evaluation and Research’s (CDER) Office of Drug Evaluation IV. A sponsor seeking to market its product OTC, either as a new drug application (NDA) or as a switch from a prescription product, applies to the DNDP, which is also responsible for the development of the OTC drug monographs.
DNDP will oversee drug development and may obtain input from the Division of Nonprescription Clinical Evaluation (DNCE) among other divisions and offices within CDER. After a sponsor submits an NDA, DNDP reviews the consumer studies, post-marketing safety data, OTC labeling, and any regulatory issues. The DNCE collaborates with DNDP and typically provides review of the efficacy and safety data related to controlled clinical trials. Additional input is obtained as needed from other disciplines outside of DNDP, including clinical pharmacology, statistics, and chemistry.
OTC drug monographs are active ingredient-based general regulations including acceptable ingredients, dosage forms, labeling, and required testing for each OTC drug class. OTC drug monographs are continually updated to add additional ingredients and labeling as needed. Updating the monographs is the primary responsibility of DNDP via the Division of Nonprescription Regulation Development (DNRD).
Although preapproval by FDA for drugs marketed under a drug monograph is not required, many companies seek assurance that the product they intend to market under the monograph complies with the regulations. These cases are primarily handled by DNDP unless consultation with the Division of Unapproved Drugs and Labeling Compliance or another review division is necessary. If a drug cannot comply with the monograph, an investigational new drug (IND) application and approved NDA are necessary before the drug product may be marketed.
FDA review divisions are assisted by advisory committees when evaluating the safety and effectiveness of OTC drug products. The Nonprescription Drugs Advisory Committee is called upon by FDA when drugs under evaluation – for switch from prescription to OTC or new drugs under development for marketing direct to consumers – present issues best evaluated by experts in the field. FDA may also ask the advisory committee to evaluate issues of safety and efficacy of monographed drugs for good cause.
Today there are more than 80 classes (therapeutic categories) of OTC drugs available to consumers without a prescription, including more than 100,000 OTC drug products encompassing about 800 active ingredients. Many of these drug products are marketed under OTC monographs and have not undergone FDA review. While this may appear as a modest oversight approach given the size and complexity of the market, it has worked remarkably well for more than 40 years.
References:
- The U.S. Food and Drug Administration Office Of Drug Evaluation IV: What we do. Available at https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm106342.htm.
- The U.S. Food and Drug Administration Office Of Drug Evaluation IV: Who we are. Available at https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm106325.htm.
- The U.S. Food and Drug Administration Bringing Nonprescription drugs to market. Available at https://www.accessdata.fda.gov/scripts/cder/training/OTC/topic3/topic3/index03.htm.
- The U.S. Food and Drug Administration Development and Regulation of OTC (nonprescription) Drug Products. Available at https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/ucm209647.htm