PDUFA after 25 Years
By Mark Land, AAHP president
July 1, 2017
The Prescription Drug User Fee Authorization (PDUFA) first enacted in 1992 was the third major piece of drug legislation in the modern era. The first two were the Drug Amendments Act of 1962 and the Drug Price Competition and Patent Term Restoration Act of 1984. PDUFA, like its predecessors, arose from overly long review times for new drug applications and a lack of transparency in the review and approval process. Unlike the predecessor legislation, PDUFA was also championed by patients awaiting life-saving drugs. Initial goals of PDUFA were simply to speed up the review process. It can be argued that the lessons learned from PDUFA I enabled later legislation that brought women, minorities, and children into the study subject populations, greatly enhancing our knowledge of disease and pharmaceutical medicine in these populations.
Why do we care about PDUFA since most homeopathic products are sold without prescription and they are not subject to user fees? There is a practical and academic answer to that question. First, in practical terms, PDUFA reauthorization sets the agenda for FDA across the drug and biologics centers. PDUFA reauthorization can be considered FDA’s drug roadmap for the following five years. PDUFA impacts our businesses through scientific thinking in the form of new guidance documents and operationally in terms of electronic systems, inspection priorities, and more. It is prudent surveillance to follow and understand PDUFA commitments. Secondly, the history of PDUFA is the history of drug development in the most recent generation. Analysis of commitments and accomplishments is a wonderful window into FDA’s strengths, weaknesses, and priorities.
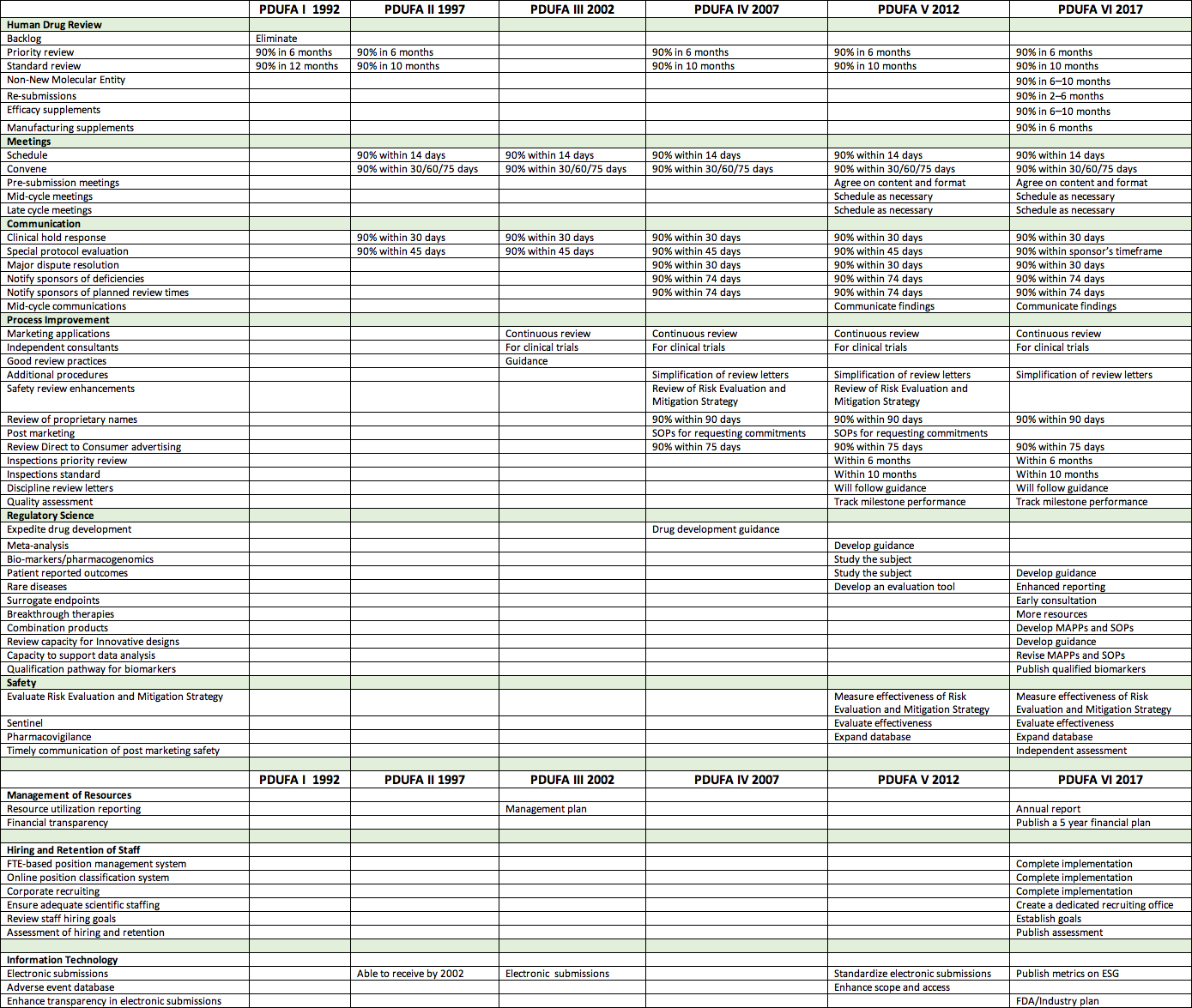
FDA commitments PDUFA I – PDUFA VI (Click to enlarge or email info@theaahp.org for a copy.)
In preparation for this article, I freshen up a review of PDUFA commitments for each of the five authorizations. In short, that review tracks the migration of FDA/industry priorities over that time. Beginning with simple approval timeframes, PDUFA now includes timeframes, communications, meetings, regulatory science, post marketing, and management commitments.
At its heart FDA is a science-driven organization. It responds well to tasks that can be measured. For example one commitment has been to finalize review of 90 percent of priority applications within six months. FDA accomplished that within two PDUFA cycles or 10 years. Considering we began in 1992 at 25 months, that was an accomplishment. Areas of greater challenge for FDA are things like electronic submissions. We are now four cycles into attempts to resolve electronic issues. Introspection is also challenging for FDA. We are now three cycles into independent assessment of performance enhancement.
Don’t get me wrong — FDA has accomplished great things in 25 years. But it still lags behind industry expectations in business and organizational management. In fairness, however, FDA drug and biologic centers have withstood a burgeoning workload. We must keep in mind that every year, 30 or so new molecular entities are approved resulting in many more individual products. As more and more drugs enter the market, FDA’s burden for review of amendments increases.