What About Your APIs and Q7 Requirements?
By Eric L. Foxman, R.Ph.
Jan. 26, 2017
In September 2016, the U.S. Food and Drug Administration (FDA) issued a guidance document, entitled Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients—Guidance for Industry. The document applies to the manufacture of Active Pharmaceutical Ingredients (APIs) for use in human drug products and closely follows a similar document published by the International Council on Harmonisation1. Does this document impact your homeopathic products? If so, to what extent? Does it matter if your company isn’t responsible for every part of the production process?
Before answering these questions, it is perhaps helpful to read the Agency’s own definition of an API starting material:
“… a raw material, an intermediate, or an API used in the production of an API…. [It] can be an article of commerce, a material purchased from one or more suppliers under contract or commercial agreement, or produced in- house…”
FDA’s Q7 Guidance covers “… APIs that are manufactured by chemical synthesis, extraction, cell culture/fermentation, recovery from natural sources, or any combination of these processes.”
Which of these apply to your homeopathic drug products? It is possible that all of them do! If your product contains a chemical, such as Kalium muriaticum, the starting material was an API before it was taken thru the homeopathic attenuation process; it was manufactured by chemical synthesis. If your product contains other substances, such as Citricum acidum, the API definition applies, for those starting materials are produced through cell culture synthesis. “Natural” starting materials, such as Lachesis and many others are recovered from natural sources. Likewise, Arnica, Pulsatilla, and the majority of the most common homeopathic substances are all extracted from natural sources. In all these instances, the “API” is the material from which the final homeopathic drug product is produced.
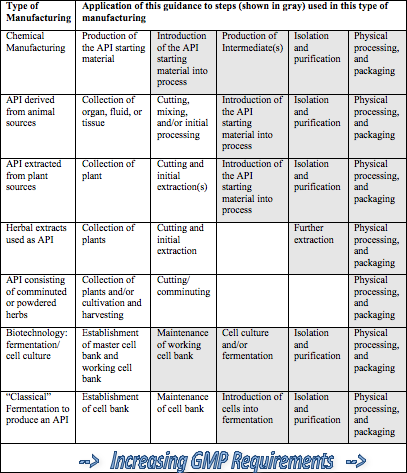
Further complicating the issue is the type of raw material that is utilized. FDA’s Q7 Guidance document identifies seven different categories of raw materials (see accompanying Table). For each of these categories, there are multiple steps in the processing — some of which the Agency considers appropriate for API Good Manufacturing Practices and some of which precede the point at which the API GMPs are applicable. The Q7 document states that each “… company should designate and document the rationale for the point at which production of the API begins….”
The burden rests firmly on each manufacturer to determine at what point the Q7 Guidelines are applicable, and these decisions need to be based on a rationale and supportable decision process. The table below (copied from FDA’s Q7 Guidance document) can help guide your decision making process. Once determined, appropriate GMPs should be applied to these intermediate and/or API manufacturing steps. The stringency of GMPs in API manufacturing should increase as the process proceeds from early API processing steps to final steps.
And yet the picture can be even more complicated than the above implies. If a company manufactures a homeopathic botanical tincture, that tincture could be a final drug product made available to consumers (e.g., Arnica Tincture). At the same time, that tincture could be used to manufacture further attenuations (e.g., Arnica 3X or 30C), at which point the tincture might be considered an API for those further attenuations. Thus, some companies may need to consider whether they will assign some kind of “dual status” to tinctures and similar early processing/attenuation steps. Alternatively, a higher level of GMP compliance can be utilized for these homeopathic drug products, thereby avoiding the complexity of dealing with categorizing and complying with two cGMP criteria.
Homeopathic marketing companies that utilize outside vendors for some or all of their manufacturing, processing and/or packaging steps are not exempt from these considerations. Written contracts with vendors should assign clear responsibilities for compliance with the appropriate level of cGMPs. In the absence of clearly defined roles and duties, marketers could find their product supply chain in jeopardy if a contract manufacturer fails to adhere to the relevant cGMPs. In fact, FDA’s Q7 Good Manufacturing Practice Guidance specifically addresses relationships and responsibilities of marketers with contract manufacturers. The recently released FDA Guidance document should be required reading for all.
Table 1 (Gray shaded cells are steps to which Q7 applies)
[1] The International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) is a project that brings together the regulatory authorities of Europe, Japan and the United States and experts from the pharmaceutical industry in the three regions to discuss scientific and technical aspects of pharmaceutical product registration. For more info, see http://www.ich.org/home.html.
Reference:
Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients–Guidance for Industry. Available at http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073497.pdf.